Abstract
The Therac-25 was a radiation machine designed to treat cancer by shining either an electron beam or X-rays into the tumorous tissue. Several design problems like software bugs and lack of hardware interlocks caused six accidents where patients received exorbitant radiation overdoses. Four of those accidents resulted in catastrophic deaths. Therac-25 manufacturers failed to acknowledge the problem and also failed to notify the Food and Drug Administration (FDA) in a timely manner, and dismissed it because they were convinced that their software was safe and capable of preventing an overdose. In the following post-mortem report, I address the issues to a third-party government oversight board for better quality control and auditing of medical equipment. I also propose several recommendations that should have been taken by Atomic Energy of Canada Limited AECL and the FDA and that now should serve as general guidelines for any new medical device that is approved in the future. Such recommendations include maintaining an audited documentation and procedure log, designing better warning and safety mechanisms, and informing the pertaining authorities and general public as soon as a problem is identified.
Problem Statement
Medical devices are developed with the goal of treating patients suffering from a particular disease. Although there always exists a risk for counter reactions and malfunctions, those are usually informed to the patient before the treatment takes place. Between 1985 and 1987, 6 people were severely injured or killed by an overdose of radiation from the Therac-25, something that was labeled as a “mistake”. That information was never really released to the public until several years later. The patients that accepted a treatment under the Therac-25 during that period were under the impression that the machine was completely safe and were excited about its highly-marketed success stories. Product failures not caught during the testing period can be costly, but failures known and not reported are fatal. The solution to this problem is to institutionalize a quality control board that oversees, revises and tests a medical product before it is released and ensures that the best design and safety practices have been followed.
Background
About the Therac-25
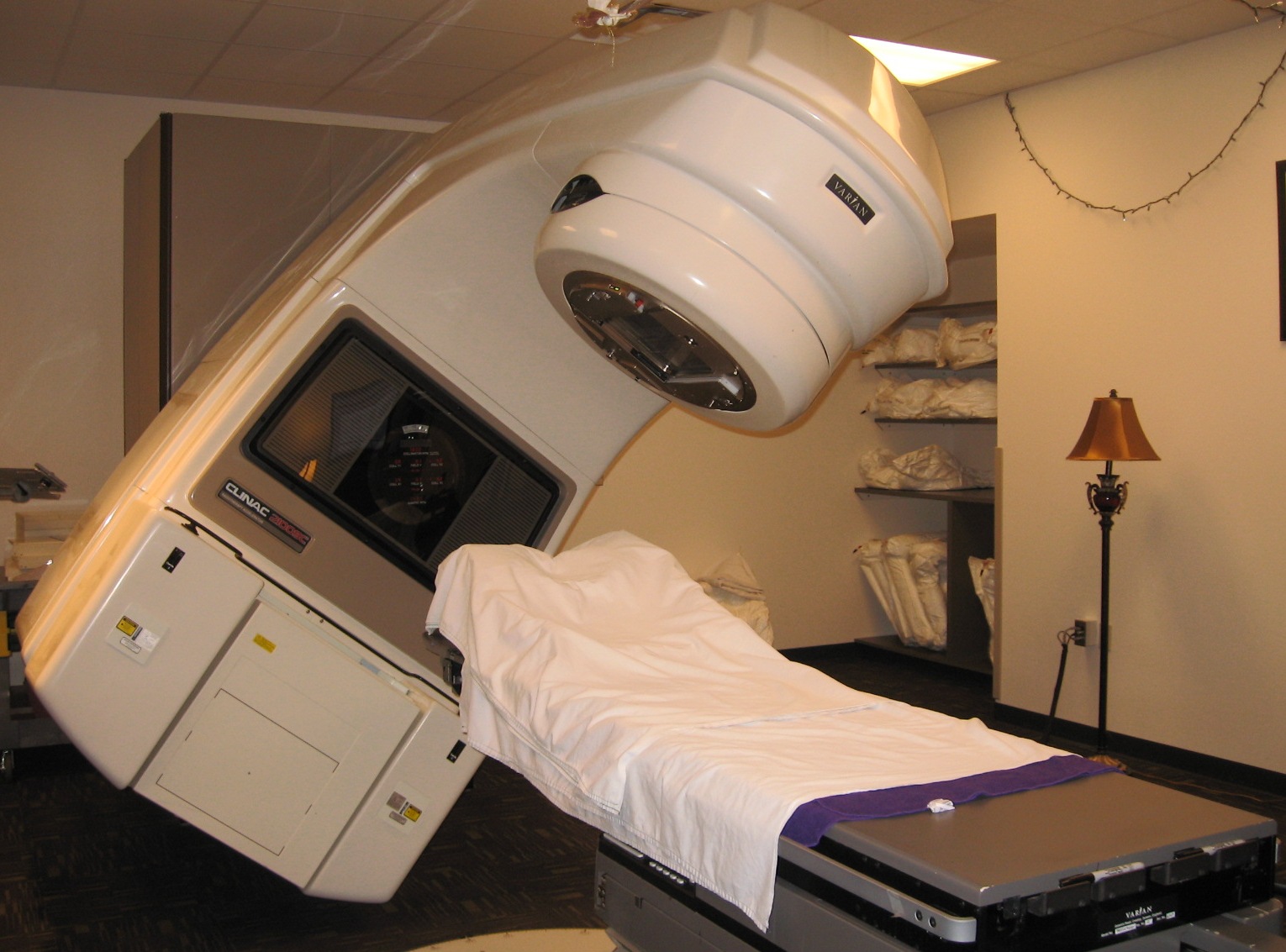
Radiotherapy is a widely-used method used to treat patients suffering from cancer. During this procedure, X-rays or a beam of electrons are shined into the tumorous cell with the goal of inhibiting any further growth of the cancerous cells while producing minimal damage to any surrounding healthy tissue (Leveson 1995). The Therac-25 (Figure 1) was a medical linear accelerator (or linac) developed by joint-venture between AECL (Atomic Energy of Canada Limited) and CGR (Compagnie Generale Radiologie) designed to treat cancerous cells using radiotherapy. When the tissue penetration needed to be shallow (i.e. surface tumors), the Therac-25 used an electron beam. For harder-to-reach tissue, the beam of electrons was transformed into X-rays using a metal plaque as shown in Figure 2 (Leveson 1993).

Figure 1: The Therac-25, a medical linear accelerator developed to treat patients suffering from cancer by shining electron beams and X-rays directly to the tumorous tissue.
The Therac-25 was the successor of the previous models Therac-6 and Therac-20. In contrast to the Therac-6 and Therac-20, which were standalone and largely hardware controlled, the Therac-25 implemented more sophisticated software and was controlled remotely by an operator in an adjacent room to protect the operator from any radiation doses (Leveson 1993). Among the medical community, there were no doubts about the capabilities of the Therac-25 to remove cancerous tissue, as “AECL Medical equipment was widely considered the best in a growing field” (Rose 1995). Patients that were recommended to the Therac-25 had already gone through a surgical procedure that extracted most of the tumor; they were scheduled for a handful of sessions to slowly and safely remove any remaining cancerous tissue that could cause regeneration of cancerous cells.
Therac-25 accidents
In total, six accidents occurred between June 1985 and January 1987 where patients received massive overdoses of radiation, leading to severe permanent injuries and four deaths (Leveson 1995). Nevertheless, the operators were under the impression that because of the ‘many safety mechanisms’ in place it was practically “impossible to overdose a patient” (Leveson 1995). During an investigation conducted, the researchers determined that two issues caused the problem. First, the software developed to control the machine failed when the machine was switched from high energy state to low energy state within 8 seconds, fully missing any necessary safety checks such as double checking that the amount of radiation that the screen showed to the operator was indeed the amount of radiation applied to the patient. Second, the hardware design neglected any physical interlocks to prevent failures (Lim 1998). Further analysis and description of the problems is provided in the following Engineering Failure section.
Engineering Failure
Therac-25’s modes of operation
There were two main modes of operation of the Therac-25 (See Figure 2):
- A low energy mode, in which a beam of electrons of 200 rads was shined directly at the patient.
- A high energy mode, at 25 MeV (million electron volts) or 25000 rads, which was shined through a metal plate before reaching the patient to transform the electron beam into X-rays (Wang 2017).

Figure 2: Two Modes of Operation of the Therac-25. One mode is shining a low energy electron beam directly at the patient. The other mode is shining the electron beam (at 25 million e- volts) through a metallic plaque to transform the beam into X-rays. The purpose of each mode is to remove cancerous cells at different tissue depths.
There are multiple factors to analyze in determining what went wrong with the Therac-25, and what caused the multiple severe injuries and even deaths to patients. The case of Ray Cox is a particularly shocking example of the harm caused by the Therac-25.
A typo that caused a death
Well-written software must be accompanied not only by well-written documentation, but also must include boundary case checks to ensure that the program behaves correctly for every input. If either of the two are lacking, then users can make fatal mistakes. On March 21, 1986, Ray Cox went to the East Texas Cancer Center for his regular radiation treatment to cure a back tumor. About 500 other patients had undergone a successful treatment with the Therac-25 at that facility during the previous two years (Rose 1994). This was Cox’s ninth session with the Therac-25 (Leveson 1995). As part of the routinary procedure, the operator placed Cox in the bed and closed the door of the room to initiate the treatment. It is important to note that that day “the intercom was broken and the video monitor was unplugged” (Rose 1994). When the operator was setting up the machine, he mistakenly typed the character “x” which indicated X-ray beam. Noticing his mistake, he quickly moved the cursor back and changed the character to an “e” for electron beam, since this was a routine and small radiation session. Eight seconds elapsed from the time the operator noticed the mistake to the time he changed it. When the beam replied with a “beam ready” message, the operator pressed “b” to administer the beam to the patient. Then, after a few seconds, the Therac-25 replied with an error showing “Malfunction 54”. The technician was used to seeing these errors which in some cases appeared up to “forty times a day” (Rose 1994), so he dismissed it and pressed “p” to proceed with the treatment. According to an FDA memorandum written after one accident, the Therac manual did not provide any information regarding any type of Malfunction errors nor did it mention that it “could place a patient at risk” (Leveson 1995). Meanwhile at the patient’s room, Cox had already been administered an X-ray radiation and was radiated again when the operator pressed “p”. Cox, instead of receiving the prescribed dose of 180 rads, received one of approximately 16000 rads or almost 90 times the dose he needed. The overdose of radiation cause a total paralysis on his left arm, both legs, vocal cords and diaphragm. He was hospitalized but died after 5 months (Leveson 1995).
The bug and the manufacturer’s response
While the operator was quick to notice his typing mistake (and correct it), the machine in the patient’s room wasn’t able to acknowledge the change as quickly. The metal plate shown in Figure 3 had moved away indicating a low energy electron beam mode, however the beam intensity administered was still in high energy X-ray mode. No communication occurred between the operator and the patient while the first 2 doses were being administered.
Figure 3: Metal plate is away from the beam indicating a low energy mode, however the Therac-25 beam intensity is still that of a high energy mode. This causes an overdose of uncontrolled radiation in the patient
The East Texas Cancer Center shut down the Therac-25 the day following the accident. Two AECL engineers spent a whole day trying to replicate the error but were not successful. The exact 8 second mistake discussed during Cox’ accident was never tried out during the testing or the investigation. The AECL engineers, after failing to reproduce the mistake, reiterated that it was impossible for the Therac-25 to overdose a patient. AECL did not inform any authority about this issue (Leveson 1995).
A deeper look into the issue with AECL’s Management
A major issue in the Therac-25 accident negligence was the inability of the AECL management team to recognize that something could be wrong with its machine. The Therac-25 was envisioned to be controlled by software, and as such the creators of the Therac-25 praised themselves by touting that the Therac-25 was controlled exclusively (and safely) by a computer. This put more emphasis in the software design to control safety than in its hardware components. Previous models had hardware and machinery to prevent accidents. In the Therac-25 everything was to be analyzed by a computer. The creators of the Therac-25 wanted to reduce development costs and production time by reducing the amount of hardware components needed, so they shifted to a more software-heavy approach which was to be completed much quicker and thus aligned with the development timeline set by its board. While the Therac-20 implemented preventive interlocks in its hardware architecture, the Therac-25 relied more heavily in its software for any prevention mechanisms. AECL took advantage of the fact that the beam intensity could be checked with a computer and decided to not duplicate the functionality with the hardware (Leveson 1995). Moreover, a bug that had already been present in the Therac-20 was found in the Therac-25. All of this combined with the fact that the manufacturers believed that the machine could not fail, resulted in AECL’s failure to acknowledge and address the cause of the failure in a timely manner.
Regulatory Problems
Any medical product that enters the market must be explicitly approved by the FDA. In the case of medical accelerators, the FDA issues a pre-market notification that determines if the product is as safe and as efficient (if not better) than the current alternatives already out there. The FDA, being reactive to problems, requires manufacturers to report serious issues (only). It is worth noting that the FDA only required equipment manufacturers and importers to report deaths and injuries but this requirement didn’t extend to health care professionals or hospitals (Felciano 1995). According to a study conducted by GAO in 1990, “the FDA knew less than 1 percent of deaths, serious injuries, or equipment malfunctions that occurred in hospitals” (Leveson 1995). Once the FDA is notified of an issue, it is responsible for issuing a corrective action plan CAP. After one of the accidents occurred, the AECL issued a voluntary recall of the Therac-25, which the FDA “termed a Class II recall” (Leveson 1995), where usage of the machine was determined that it could cause “temporary or medically reversible consequences or where the probability of serious adverse health consequences is remote” (Leveson 1995). The FDA followed the recall closely and after AECL made the modifications to the Therac-25, they told users that they “could return to normal operating procedures” (Leveson 1995). Clearly the FDA did not revise the specifications and most importantly did not understand the implications of the accidents at the time. The approved CAP by the FDA was never fully completed until 2 years later and more importantly, most of the changes were not related to improving the safety of the machine itself. (Rose 1994)
Ethical Analysis
To analyze whether the management, technical and regulatory actions taken regarding the Therac-25 accidents were justified, I will apply Kantianism as my ethical framework. Kantianism was an ethical philosophy developed by Immanuel Kant, a German theorist. Kantianism essentially poses the following two questions and guides our decisions as to how to act ethically. The first question is whether or not we can will that everyone act as one proposes to act (universality principle) and the second question is whether or not the actions taken respect the goals of the human beings involved rather than using them as means to the end (reciprocity principle). If either answer to either question is negative, then we must not perform the action. A categorical imperative is an action that must be performed unconditionally. Kantianism analyzes actions from a deontological standpoint, which evaluates an action in accordance to a particular moral rule.
Having defined Kantianism, we can now apply it to the Therac-25 case. To apply Kant’s first categorical imperative, the universality principle, we must first analyze whether AECL’s maxim can be universalized and willed without contradiction. The specific action performed by AECL was to ignore the safety failures produced by the Therac-25 because they were unable to reproduce them during the non-conclusive investigation phase. In this case, AECL’s maxim is “AECL won’t instruct Therac-25 users to stop using the machine despite being aware of deadly accidents of which causes haven’t been determined”. The universal law would then become “Any FDA-approved machine shall continue its operation as long as no accident can be replicated during the inspection process”. If this were the case, then no patient would be willing to undergo treatment because they wouldn’t be able to trust the safety of the machine or the ability of the auditor to conduct a proper test. Therefore, this maxim can’t be universalized and hence should not be followed by AECL.
AECL also violated Kant’s second categorical imperative, the reciprocity principle, by placing the patient’s’ safety after their own interests. AECL only analyzed its self-interests in terms of public image and decided to not inform their clients about the accidents for a long time, thus regarding their patients as means to an end. The goal of the machine had shifted from treating patients to not damaging the public image of the company. From this, it follows that AECL should have made the accidents public so that patients would know the risks involved when submitted to a treatment under the Therac-25 and so they could be in the capacity of making an autonomous rational decision for themselves. In addition to that, AECL failed in their duty of ensuring public safety, which they claim on the mission statement to be a core value in their decisions. It was immoral to not address the situation quickly before it took away so many lives.
Recommendations
The Health Protection Branch of the Canadian government and United States Food and Drug Administration (FDA) investigated the issue regarding the malfunctioning of the Therac-25. On February 10, 1987, they deemed it unsafe to use and shut it down until further notice. On July 21, 1987 AECL provided the following recommendations to deal with the issue (Lim 1998):
- If the machine restarts, operators must re-enter patient information and machine settings
- Operators must check that the metal plate is in the correct place if the X-ray mode is selected
- The exact dose amount will be shown to the operator on the screen
- Operators manuals must be re-written to reflect the new changes
- The editing keys capabilities be limited to prevent any accidental typos
As the governing body that should oversee medical equipment introductions, it is imperative that you ensure that these recommendations are followed. Expanding on those recommendations, here are further suggestions.
On software production procedures and auditing
Software creation for real-time systems is not only time consuming and complicated but it also is “the most challenging and complex task that can be undertaken by a software engineer” (Wang 2017). The software used in the Therac-25 was expanded upon that of the Therac-20 which in turn was developed from the Therac-6. One programmer (over the years) was in charge of revising the portability of the software. Strict software revision procedure must be put in place to ensure that code sent to production is as bug-free as possible. If there are any concerns of possible failure for a particular subset of the code, this code must be sent back to testing immediately and any components that use it must be temporarily halted. As the oversight committee, you should enforce the aforementioned software development procedure for any company that is producing medical software to ensure that only the best software practices are followed. Software designers should create clear and locking warning messages when any software or hardware errors occur (Gowen 1994). Any legacy software should be thoroughly tested when adapted for a new piece of equipment. There must be at least two people to revise any software that is being imported from an external source. A person should sign off any changes or major decisions made regarding software functionality. (Felciano 1995) Financial and legal actions should be pursued against any companies that fail to comply.
On Investigations
When trying to reproduce mistakes, the manufacturers of the device should use the log of the actions performed by the operator in the same exact sequence and timing to ensure that the same conditions that caused the error are met. All investigations must also be logged and audited by the regulating body, which must be present during any critical investigation. The manufacturer should err on the side of safety and discontinue the use of a machine if its safety is compromised. The manufacturer should not believe the machine is error-free from the start. A recall and public message must be issued to all patients immediately after a mistake is found. Moreover, a period investigation must be conducted to a controlled set of machines to check that hospitals are also complying with the usage recommendations set forth by the manufacturer.
On the creation of a third-party body
Additionally, a regulating body (a capable doctor or technician not related to the company) must be installed to conduct the audit, rather than having the company self-check a mistake. If a critical mistake is found, AECL and the regulating body must report the error to all the hospitals and also to the FDA. This double-check ensures that everyone is held accountable. In the case of the FDA, they should (as they have been implementing over the years) set up an evaluation procedure in place where stricter and more thorough control for any new medical equipment. They should also require any equipment manufacturer to notify them immediately in case of any problems. An unbiased and unrelated regulating body can at a minimum provide an extra layer of security and procedure-compliance to ensure that medical devices meet the necessary requirements throughout its lifetime, not just during its marketing and development.
Conclusion
The Therac-25 history is an interesting case of bad software practices, negligence and failure to acknowledge and divulge problems. 6 people were injured or killed due to poor testing and precarious error checks. That is six people too many. AECL’s self interests in keeping a good image and not becoming involved in larger lawsuits should not have been able to impact a patient’s health. Drawing from this case, the FDA now requires any medical software to comply with a documentation standard in which decisions and instructions are detailed so they can be traced back by a third-party committee if any problems arise. In January of 1995, the International Electrotechnical Commission recommended “software safety standards for medical equipment, standards developed partly as a result of the Therac-25 accidents” (Rose 1994). While engineers in recent years have experienced a drop in their productivity due to documentation requirements, this measure is intended to reduce the bugs that are pushed out live to production (Rose 1994). The Therac-25 case made it clear that such requirements are imperative to ensure good software quality control.
References
Felciano, R.M. Human Error: Designing for Error in Medical Information Systems. Stanford University School of Medicine 1995: 1-6.
Leveson, N., Turner, C.S. “An Investigation of the Therac-25 Accidents”. Reprinted from IEEE
Computer, Vol.26, No.7, July 1993, pp.18-41. http://ei.cs.vt.edu/~cs3604/lib/Therac_25/Therac_1.html [Accessed Jan 14th 2017]
Leveson, N. “Medical Devices: The Therac-25” in N. Leveson, Safeware. System, Safety and Computers, Addison-Wesley. 1995.
http://sunnyday.mit.edu/papers/therac.pdf [Accessed Jan 15th 2017]
Lim, J. “An Engineering Disaster. Therac-25” Bowdoin 1998 http://www.bowdoin.edu/~allen/courses/cs260/readings/therac.pdf [Accessed Jan 18th 2017]
Rose, B. “Fatal Dose. Radiation Deaths linked to AECL Computer Errors.” Canadian Coalition for Nuclear Responsibility. 1994.
Wang, J. “Therac-25 and industrial design engineering of socio-technical systems” in Wang, J. Industrial Design Engineering: Inventive Problem Solving, CRC Press 2017.
https://www.crcpress.com/authors/news/i3158-therac-25-and-industrial-design-engineering-of-socio-technical-systems [Accessed Jan 20th 2017]
Therac-25 Post-Mortem Report
Abstract
The Therac-25 was a radiation machine designed to treat cancer by shining either an electron beam or X-rays into the tumorous tissue. Several design problems like software bugs and lack of hardware interlocks caused six accidents where patients received exorbitant radiation overdoses. Four of those accidents resulted in catastrophic deaths. Therac-25 manufacturers failed to acknowledge the problem and also failed to notify the Food and Drug Administration (FDA) in a timely manner, and dismissed it because they were convinced that their software was safe and capable of preventing an overdose. In the following post-mortem report, I address the issues to a third-party government oversight board for better quality control and auditing of medical equipment. I also propose several recommendations that should have been taken by Atomic Energy of Canada Limited AECL and the FDA and that now should serve as general guidelines for any new medical device that is approved in the future. Such recommendations include maintaining an audited documentation and procedure log, designing better warning and safety mechanisms, and informing the pertaining authorities and general public as soon as a problem is identified.
Problem Statement
Medical devices are developed with the goal of treating patients suffering from a particular disease. Although there always exists a risk for counter reactions and malfunctions, those are usually informed to the patient before the treatment takes place. Between 1985 and 1987, 6 people were severely injured or killed by an overdose of radiation from the Therac-25, something that was labeled as a “mistake”. That information was never really released to the public until several years later. The patients that accepted a treatment under the Therac-25 during that period were under the impression that the machine was completely safe and were excited about its highly-marketed success stories. Product failures not caught during the testing period can be costly, but failures known and not reported are fatal. The solution to this problem is to institutionalize a quality control board that oversees, revises and tests a medical product before it is released and ensures that the best design and safety practices have been followed.
Background
About the Therac-25
Radiotherapy is a widely-used method used to treat patients suffering from cancer. During this procedure, X-rays or a beam of electrons are shined into the tumorous cell with the goal of inhibiting any further growth of the cancerous cells while producing minimal damage to any surrounding healthy tissue (Leveson 1995). The Therac-25 (Figure 1) was a medical linear accelerator (or linac) developed by joint-venture between AECL (Atomic Energy of Canada Limited) and CGR (Compagnie Generale Radiologie) designed to treat cancerous cells using radiotherapy. When the tissue penetration needed to be shallow (i.e. surface tumors), the Therac-25 used an electron beam. For harder-to-reach tissue, the beam of electrons was transformed into X-rays using a metal plaque as shown in Figure 2 (Leveson 1993).
Figure 1: The Therac-25, a medical linear accelerator developed to treat patients suffering from cancer by shining electron beams and X-rays directly to the tumorous tissue.
The Therac-25 was the successor of the previous models Therac-6 and Therac-20. In contrast to the Therac-6 and Therac-20, which were standalone and largely hardware controlled, the Therac-25 implemented more sophisticated software and was controlled remotely by an operator in an adjacent room to protect the operator from any radiation doses (Leveson 1993). Among the medical community, there were no doubts about the capabilities of the Therac-25 to remove cancerous tissue, as “AECL Medical equipment was widely considered the best in a growing field” (Rose 1995). Patients that were recommended to the Therac-25 had already gone through a surgical procedure that extracted most of the tumor; they were scheduled for a handful of sessions to slowly and safely remove any remaining cancerous tissue that could cause regeneration of cancerous cells.
Therac-25 accidents
In total, six accidents occurred between June 1985 and January 1987 where patients received massive overdoses of radiation, leading to severe permanent injuries and four deaths (Leveson 1995). Nevertheless, the operators were under the impression that because of the ‘many safety mechanisms’ in place it was practically “impossible to overdose a patient” (Leveson 1995). During an investigation conducted, the researchers determined that two issues caused the problem. First, the software developed to control the machine failed when the machine was switched from high energy state to low energy state within 8 seconds, fully missing any necessary safety checks such as double checking that the amount of radiation that the screen showed to the operator was indeed the amount of radiation applied to the patient. Second, the hardware design neglected any physical interlocks to prevent failures (Lim 1998). Further analysis and description of the problems is provided in the following Engineering Failure section.
Engineering Failure
Therac-25’s modes of operation
There were two main modes of operation of the Therac-25 (See Figure 2):
- A low energy mode, in which a beam of electrons of 200 rads was shined directly at the patient.
- A high energy mode, at 25 MeV (million electron volts) or 25000 rads, which was shined through a metal plate before reaching the patient to transform the electron beam into X-rays (Wang 2017).
Figure 2: Two Modes of Operation of the Therac-25. One mode is shining a low energy electron beam directly at the patient. The other mode is shining the electron beam (at 25 million e- volts) through a metallic plaque to transform the beam into X-rays. The purpose of each mode is to remove cancerous cells at different tissue depths.
There are multiple factors to analyze in determining what went wrong with the Therac-25, and what caused the multiple severe injuries and even deaths to patients. The case of Ray Cox is a particularly shocking example of the harm caused by the Therac-25.
A typo that caused a death
Well-written software must be accompanied not only by well-written documentation, but also must include boundary case checks to ensure that the program behaves correctly for every input. If either of the two are lacking, then users can make fatal mistakes. On March 21, 1986, Ray Cox went to the East Texas Cancer Center for his regular radiation treatment to cure a back tumor. About 500 other patients had undergone a successful treatment with the Therac-25 at that facility during the previous two years (Rose 1994). This was Cox’s ninth session with the Therac-25 (Leveson 1995). As part of the routinary procedure, the operator placed Cox in the bed and closed the door of the room to initiate the treatment. It is important to note that that day “the intercom was broken and the video monitor was unplugged” (Rose 1994). When the operator was setting up the machine, he mistakenly typed the character “x” which indicated X-ray beam. Noticing his mistake, he quickly moved the cursor back and changed the character to an “e” for electron beam, since this was a routine and small radiation session. Eight seconds elapsed from the time the operator noticed the mistake to the time he changed it. When the beam replied with a “beam ready” message, the operator pressed “b” to administer the beam to the patient. Then, after a few seconds, the Therac-25 replied with an error showing “Malfunction 54”. The technician was used to seeing these errors which in some cases appeared up to “forty times a day” (Rose 1994), so he dismissed it and pressed “p” to proceed with the treatment. According to an FDA memorandum written after one accident, the Therac manual did not provide any information regarding any type of Malfunction errors nor did it mention that it “could place a patient at risk” (Leveson 1995). Meanwhile at the patient’s room, Cox had already been administered an X-ray radiation and was radiated again when the operator pressed “p”. Cox, instead of receiving the prescribed dose of 180 rads, received one of approximately 16000 rads or almost 90 times the dose he needed. The overdose of radiation cause a total paralysis on his left arm, both legs, vocal cords and diaphragm. He was hospitalized but died after 5 months (Leveson 1995).
The bug and the manufacturer’s response
While the operator was quick to notice his typing mistake (and correct it), the machine in the patient’s room wasn’t able to acknowledge the change as quickly. The metal plate shown in Figure 3 had moved away indicating a low energy electron beam mode, however the beam intensity administered was still in high energy X-ray mode. No communication occurred between the operator and the patient while the first 2 doses were being administered.
Figure 3: Metal plate is away from the beam indicating a low energy mode, however the Therac-25 beam intensity is still that of a high energy mode. This causes an overdose of uncontrolled radiation in the patient
The East Texas Cancer Center shut down the Therac-25 the day following the accident. Two AECL engineers spent a whole day trying to replicate the error but were not successful. The exact 8 second mistake discussed during Cox’ accident was never tried out during the testing or the investigation. The AECL engineers, after failing to reproduce the mistake, reiterated that it was impossible for the Therac-25 to overdose a patient. AECL did not inform any authority about this issue (Leveson 1995).
A deeper look into the issue with AECL’s Management
A major issue in the Therac-25 accident negligence was the inability of the AECL management team to recognize that something could be wrong with its machine. The Therac-25 was envisioned to be controlled by software, and as such the creators of the Therac-25 praised themselves by touting that the Therac-25 was controlled exclusively (and safely) by a computer. This put more emphasis in the software design to control safety than in its hardware components. Previous models had hardware and machinery to prevent accidents. In the Therac-25 everything was to be analyzed by a computer. The creators of the Therac-25 wanted to reduce development costs and production time by reducing the amount of hardware components needed, so they shifted to a more software-heavy approach which was to be completed much quicker and thus aligned with the development timeline set by its board. While the Therac-20 implemented preventive interlocks in its hardware architecture, the Therac-25 relied more heavily in its software for any prevention mechanisms. AECL took advantage of the fact that the beam intensity could be checked with a computer and decided to not duplicate the functionality with the hardware (Leveson 1995). Moreover, a bug that had already been present in the Therac-20 was found in the Therac-25. All of this combined with the fact that the manufacturers believed that the machine could not fail, resulted in AECL’s failure to acknowledge and address the cause of the failure in a timely manner.
Regulatory Problems
Any medical product that enters the market must be explicitly approved by the FDA. In the case of medical accelerators, the FDA issues a pre-market notification that determines if the product is as safe and as efficient (if not better) than the current alternatives already out there. The FDA, being reactive to problems, requires manufacturers to report serious issues (only). It is worth noting that the FDA only required equipment manufacturers and importers to report deaths and injuries but this requirement didn’t extend to health care professionals or hospitals (Felciano 1995). According to a study conducted by GAO in 1990, “the FDA knew less than 1 percent of deaths, serious injuries, or equipment malfunctions that occurred in hospitals” (Leveson 1995). Once the FDA is notified of an issue, it is responsible for issuing a corrective action plan CAP. After one of the accidents occurred, the AECL issued a voluntary recall of the Therac-25, which the FDA “termed a Class II recall” (Leveson 1995), where usage of the machine was determined that it could cause “temporary or medically reversible consequences or where the probability of serious adverse health consequences is remote” (Leveson 1995). The FDA followed the recall closely and after AECL made the modifications to the Therac-25, they told users that they “could return to normal operating procedures” (Leveson 1995). Clearly the FDA did not revise the specifications and most importantly did not understand the implications of the accidents at the time. The approved CAP by the FDA was never fully completed until 2 years later and more importantly, most of the changes were not related to improving the safety of the machine itself. (Rose 1994)
Ethical Analysis
To analyze whether the management, technical and regulatory actions taken regarding the Therac-25 accidents were justified, I will apply Kantianism as my ethical framework. Kantianism was an ethical philosophy developed by Immanuel Kant, a German theorist. Kantianism essentially poses the following two questions and guides our decisions as to how to act ethically. The first question is whether or not we can will that everyone act as one proposes to act (universality principle) and the second question is whether or not the actions taken respect the goals of the human beings involved rather than using them as means to the end (reciprocity principle). If either answer to either question is negative, then we must not perform the action. A categorical imperative is an action that must be performed unconditionally. Kantianism analyzes actions from a deontological standpoint, which evaluates an action in accordance to a particular moral rule.
Having defined Kantianism, we can now apply it to the Therac-25 case. To apply Kant’s first categorical imperative, the universality principle, we must first analyze whether AECL’s maxim can be universalized and willed without contradiction. The specific action performed by AECL was to ignore the safety failures produced by the Therac-25 because they were unable to reproduce them during the non-conclusive investigation phase. In this case, AECL’s maxim is “AECL won’t instruct Therac-25 users to stop using the machine despite being aware of deadly accidents of which causes haven’t been determined”. The universal law would then become “Any FDA-approved machine shall continue its operation as long as no accident can be replicated during the inspection process”. If this were the case, then no patient would be willing to undergo treatment because they wouldn’t be able to trust the safety of the machine or the ability of the auditor to conduct a proper test. Therefore, this maxim can’t be universalized and hence should not be followed by AECL.
AECL also violated Kant’s second categorical imperative, the reciprocity principle, by placing the patient’s’ safety after their own interests. AECL only analyzed its self-interests in terms of public image and decided to not inform their clients about the accidents for a long time, thus regarding their patients as means to an end. The goal of the machine had shifted from treating patients to not damaging the public image of the company. From this, it follows that AECL should have made the accidents public so that patients would know the risks involved when submitted to a treatment under the Therac-25 and so they could be in the capacity of making an autonomous rational decision for themselves. In addition to that, AECL failed in their duty of ensuring public safety, which they claim on the mission statement to be a core value in their decisions. It was immoral to not address the situation quickly before it took away so many lives.
Recommendations
The Health Protection Branch of the Canadian government and United States Food and Drug Administration (FDA) investigated the issue regarding the malfunctioning of the Therac-25. On February 10, 1987, they deemed it unsafe to use and shut it down until further notice. On July 21, 1987 AECL provided the following recommendations to deal with the issue (Lim 1998):
- If the machine restarts, operators must re-enter patient information and machine settings
- Operators must check that the metal plate is in the correct place if the X-ray mode is selected
- The exact dose amount will be shown to the operator on the screen
- Operators manuals must be re-written to reflect the new changes
- The editing keys capabilities be limited to prevent any accidental typos
As the governing body that should oversee medical equipment introductions, it is imperative that you ensure that these recommendations are followed. Expanding on those recommendations, here are further suggestions.
On software production procedures and auditing
Software creation for real-time systems is not only time consuming and complicated but it also is “the most challenging and complex task that can be undertaken by a software engineer” (Wang 2017). The software used in the Therac-25 was expanded upon that of the Therac-20 which in turn was developed from the Therac-6. One programmer (over the years) was in charge of revising the portability of the software. Strict software revision procedure must be put in place to ensure that code sent to production is as bug-free as possible. If there are any concerns of possible failure for a particular subset of the code, this code must be sent back to testing immediately and any components that use it must be temporarily halted. As the oversight committee, you should enforce the aforementioned software development procedure for any company that is producing medical software to ensure that only the best software practices are followed. Software designers should create clear and locking warning messages when any software or hardware errors occur (Gowen 1994). Any legacy software should be thoroughly tested when adapted for a new piece of equipment. There must be at least two people to revise any software that is being imported from an external source. A person should sign off any changes or major decisions made regarding software functionality. (Felciano 1995) Financial and legal actions should be pursued against any companies that fail to comply.
On Investigations
When trying to reproduce mistakes, the manufacturers of the device should use the log of the actions performed by the operator in the same exact sequence and timing to ensure that the same conditions that caused the error are met. All investigations must also be logged and audited by the regulating body, which must be present during any critical investigation. The manufacturer should err on the side of safety and discontinue the use of a machine if its safety is compromised. The manufacturer should not believe the machine is error-free from the start. A recall and public message must be issued to all patients immediately after a mistake is found. Moreover, a period investigation must be conducted to a controlled set of machines to check that hospitals are also complying with the usage recommendations set forth by the manufacturer.
On the creation of a third-party body
Additionally, a regulating body (a capable doctor or technician not related to the company) must be installed to conduct the audit, rather than having the company self-check a mistake. If a critical mistake is found, AECL and the regulating body must report the error to all the hospitals and also to the FDA. This double-check ensures that everyone is held accountable. In the case of the FDA, they should (as they have been implementing over the years) set up an evaluation procedure in place where stricter and more thorough control for any new medical equipment. They should also require any equipment manufacturer to notify them immediately in case of any problems. An unbiased and unrelated regulating body can at a minimum provide an extra layer of security and procedure-compliance to ensure that medical devices meet the necessary requirements throughout its lifetime, not just during its marketing and development.
Conclusion
The Therac-25 history is an interesting case of bad software practices, negligence and failure to acknowledge and divulge problems. 6 people were injured or killed due to poor testing and precarious error checks. That is six people too many. AECL’s self interests in keeping a good image and not becoming involved in larger lawsuits should not have been able to impact a patient’s health. Drawing from this case, the FDA now requires any medical software to comply with a documentation standard in which decisions and instructions are detailed so they can be traced back by a third-party committee if any problems arise. In January of 1995, the International Electrotechnical Commission recommended “software safety standards for medical equipment, standards developed partly as a result of the Therac-25 accidents” (Rose 1994). While engineers in recent years have experienced a drop in their productivity due to documentation requirements, this measure is intended to reduce the bugs that are pushed out live to production (Rose 1994). The Therac-25 case made it clear that such requirements are imperative to ensure good software quality control.
References
Felciano, R.M. Human Error: Designing for Error in Medical Information Systems. Stanford University School of Medicine 1995: 1-6.
Leveson, N., Turner, C.S. “An Investigation of the Therac-25 Accidents”. Reprinted from IEEE
Computer, Vol.26, No.7, July 1993, pp.18-41. http://ei.cs.vt.edu/~cs3604/lib/Therac_25/Therac_1.html [Accessed Jan 14th 2017]
Leveson, N. “Medical Devices: The Therac-25” in N. Leveson, Safeware. System, Safety and Computers, Addison-Wesley. 1995.
http://sunnyday.mit.edu/papers/therac.pdf [Accessed Jan 15th 2017]
Lim, J. “An Engineering Disaster. Therac-25” Bowdoin 1998 http://www.bowdoin.edu/~allen/courses/cs260/readings/therac.pdf [Accessed Jan 18th 2017]
Rose, B. “Fatal Dose. Radiation Deaths linked to AECL Computer Errors.” Canadian Coalition for Nuclear Responsibility. 1994.
Wang, J. “Therac-25 and industrial design engineering of socio-technical systems” in Wang, J. Industrial Design Engineering: Inventive Problem Solving, CRC Press 2017.
https://www.crcpress.com/authors/news/i3158-therac-25-and-industrial-design-engineering-of-socio-technical-systems [Accessed Jan 20th 2017]


